Machine learning for simulation of defects in van der Waals heterostructures
Machine learning for simulation of defects in van der Waals heterostructures
Research questions:
Mono- and few-layered nano-sheets made of transition metal dichalcogenides (TMDs) are versatile materials with many potential applications ranging from energy storage to catalysis and lubrication [1]. The electronic, optical and mechanical properties of TMDs, however, are strongly affected by defects such as vacancies, rotated bonds and grain boundaries [2]. In this project, we will use atomistic simulations to study the formation, migration and stability of defects in TMD/graphene heterostructures. Our investigations complement the electron microscopy studies carried out in the project of J. Kotakoski, who will investigate defects in TMD/graphene heterostructures created by ion and electron irradiation. Ab initio trained neural network for energy and force calculations.
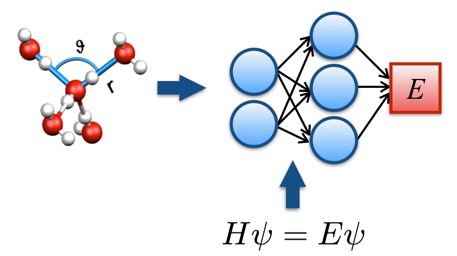
Methods:
In principle, atomistic simulations of TMDs can be performed using ab initio methods to compute forces and energies [2]. However, such calculations are computationally challenging due to the wide ranges of time and length scales characterizing the structure and dynamics of defects. Here we propose to overcome these limitations by using neural network potentials to encode ab initio potential energy surfaces (see figure). In this machine learning approach, which delivers the accuracy of density functional theory at a fraction of its cost, a high-dimensional neural network is trained using a set of high-precision reference data to predict the energies and forces required in atomistic simulations [3]. In our group, we have recently developed a neural network framework and implemented it into a popular molecular dynamics package. We will develop a new neural network model for the TMDs studied experimentally by our DCAFM colleagues and combine it with advanced rare event techniques [4] to study the processes of defect creation and migration in TMDs.
Time frame:
Months 1-9: generation of ab initio training data; months 10-12: training of neural network potential; months 13-27: MD simulations of defect conformation and migration; months 28-42: investigate free energetics of defects; months 43-48: writing of papers and thesis.
Participating DCAFM-faculty:
C. Dellago (PI), J. Kotakoski (experiments on structure and dynamics of defects in TMDs), C. Franchini (ab initio training set for machine learning).
[1] M. Chhowalla et al., Nat. Chem. 5, 263 (2013), DOI: 10.1038/nchem.1589.
[2] T. Heine, Acc. Chem. Res. 48, 65-72 (2015), DOI: 10.1021/ar500277z.
[3] T. Morawietz et al., Proc. Natl. Acad. Sci. USA 113, 8368 (2016), DOI: 10.1073/pnas.1602375113.
[4] P. G. Bolhuis and C. Dellago, Eur. Phys. J. ST 224, 1951 (2015), DOI: 10.1140/epjst/e2015-02419-6.